
The Neuroscience of Ketamine
Making the decision to embark on a ketamine therapy program is exciting, it’s a major decision for yourself and everyone you interact with. As you explore the world of ketamine therapy further, you may come across some terms and phrases you may not have encountered before — this can lead to some confusion, anxiety, and uncertainty if you’re have to make decisions for your health and wellbeing without a deeper sense of what you’re working with or how the process unfolds for you.
That is what this Glossary resource will take care of for you. It is a glossary of the common terms that may arise throughout the course of a ketamine therapy protocol, and what they mean in context and for you specifically. This is an evolving resource, it will continue to be updated as new terms arise or through the questions we receive from clients at Mindbloom®. With that being said, this is not an exhaustive list, some clinicians or other contexts may use language that is not universally shared by the community.
It’s important to ask for clarification if you need any, to investigate deeply the programs and protocols you will be working with so that you can make informed decisions for yourself and for your overall health and wellbeing.
If you have a term to suggest for this list, or would like clarification on anything listed below, feel free to reach out to our team. If you’d like to work with experts in this field familiar with the protocols, phrasing, and process of ketamine therapy, you can sign up here to start the process.
Neurons
The neuron is the fundamental cell of the brain. Each neuron has a cell body (soma) that contains the nucleus, where the cell’s DNA is located; the nucleus regulates all the processes in the neuron. The cell body also contains other organelles, such as ribosomes, needed to make proteins such as neurotransmitters, and mitochondria, the miniature power plants that provide energy in the cell.
Leading off the cell body are two specialized extensions. At one end is the axon, a single long, thin nerve fiber that transmits messages from the body of the neuron to the dendrites of other neurons. Branching off from the neuron at the other end are the dendrites, a dense, bushy cluster of nerve fibers that receive messages from the axons of other neurons and send them on to the cell body. The dendrites branch out to form a large surface area that connects with many nearby neurons.
In the cerebral cortex alone, the most highly developed part of the brain, you have somewhere between 14 and 16 billion neurons. Every moment of every day, all those neurons are making billions of connections with each other, firing together to form pathways involved in planning, learning, memory, speech, and emotion.
The Role of Neurotransmitters
Neurons communicate with each other by passing impulses (also called action potentials) from the axon of one neuron to the dendrites of another. The junction where an axon meets a dendrite is called the synapse. The axons and dendrites don’t actually touch, however—a tiny gap called the synaptic cleft separates them.
Getting the nerve impulse across the synaptic cleft is the job of neurotransmitters: chemicals that act as messengers between neurons. Neurotransmitters are manufactured in the neurons and stored in tiny sacs called vesicles at the tips of axons.
The cell membranes of neurons are studded with receptors that are binding sites for specific neurotransmitters. Each receptor is a precise match for that particular chemical messenger. The neurotransmitters attach to the receptors, fitting like keys into locks and opening channels into the cell.
When a nerve impulse travels down the axon and reaches the very end, it makes calcium channels in the cell open. The positively charged calcium ions flow in and make the synaptic vesicles fuse with the cell membrane (exocytosis). The neurotransmitters stored in the vesicles then pour into the synaptic cleft. They diffuse across the gap and bind to their matching receptors in the membrane of the postsynaptic neuron (the target neuron). The binding opens the channels leading into the cell interior and calcium, sodium, magnesium, and other ions flow in.
The flow of incoming ions such as calcium and sodium depolarizes the cell membrane, creating less negative charge inside the cell. If the depolarization threshold for making the neuron fire is reached, the impulse is transmitted through the cell, down its axon, and on to the next neuron in the chain.
What happens to the neurotransmitters once they’ve done their job of opening the channels? Your body is frugal and efficient. Reuptake receptors on the presynaptic (sending) cell membrane capture the used neurotransmitters and carry them back into the cell, where they can be repackaged into new synaptic vesicles to await another cycle.
When the neurotransmitters are re-absorbed, the synapse turns off. The whole process of shuttling an impulse through one neuron and on to the next takes only about seven milliseconds. It’s happening in your brain billions of times every second, around the clock.
Types of Neurotransmitters
Different neurotransmitters affect neurons in different ways. Excitatory transmitters excite the postsynaptic neurons, making the neuron more likely to fire an impulse. Inhibitory transmitters slow the neuron, making it less likely to fire an impulse. And just to confuse things, the neurotransmitter dopamine can be both excitatory and inhibitory, depending on which receptors are present.
In the brain, the neurotransmitters glutamate and dopamine are excitatory. The inhibitory neurotransmitters are GABA (gamma-aminobutyric acid), serotonin, and dopamine (sometimes).
The release, removal, and reuptake of neurotransmitters is tightly regulated. Too much or too little of any excitatory or inhibitory neurotransmitter, or being too sensitive or insensitive to them, can disrupt the balance.
Glutamate is the most common excitatory neurotransmitter in the brain. It plays a particularly important role in neuroplasticity (the brain’s ability to form new synapses and neural connections over a lifetime), learning, and forming memories. When there’s too much glutamate in the brain, the postsynaptic neurons can become hyperexcited; when there’s way too much glutamate in the brain, it can damage neurons or even cause neuron death.
Stress, Glutamate, and Damaged Neurons
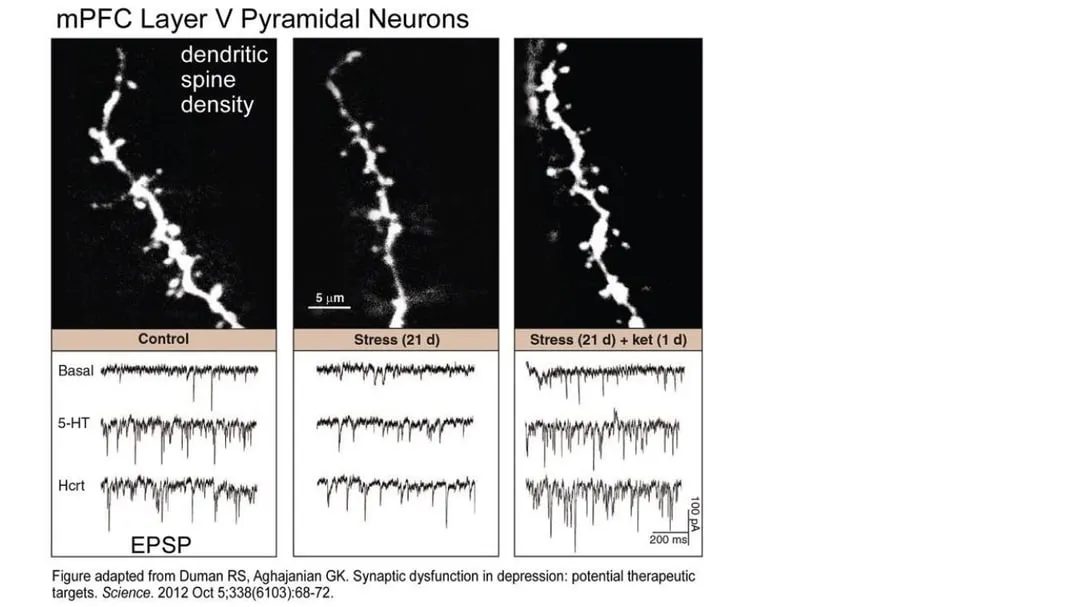
Long-lasting stress takes a toll on the neurons of the cortex. Constant high levels of the stress hormone cortisol can make neurons atrophy—they shrivel and shrink. The dendrites go from large numbers of dense, spreading branches to smaller numbers of shorter, stubby branches that make fewer connections to other neurons. The axons shrink and get thinner. Intense stress can also change glutamate signaling and make the neurons less responsive and less able to connect with other neurons. Brain imaging shows that in depressed people, the prefrontal cortex is reduced in size. Shriveled connections in the cortex mean the neural pathways that control memory, decision-making, emotions, and attention don’t work as well.
The end result of all these glutamate-related neuronal changes is a brain much more predisposed to manifesting the constellation of subjective phenomena we call depression and anxiety.
This is where ketamine comes in. The drug company researchers who developed ketamine as an anesthetic in the 1960s knew it worked by triggering glutamate release into the synapse. Later researchers studying the roots of depression discovered that in people with depression, something was going wrong in the glutamate receptors.
By the 1990s, they knew enough to start looking for a drug that would target the glutamate system. They didn’t have to look far. Ketamine, well studied by that point for its effects on neurons, had already been accidentally found to lift depression. Researchers at Yale began using very small doses of ketamine to treat people with severe depression who weren’t helped by standard antidepressant drugs. The results were startlingly successful.
Ketamine and Glutamate
Research into the full pharmacological action of ketamine is revealing just how complex the response to this drug is. We know that ketamine also modulates additional receptor types, but the glutamate receptors seem to play the most important role.
Neurons have a number of different binding sites for glutamate, but when it comes to ketamine, two are of particular interest: the NMDA (N-methyl-D-aspartate) receptor and the AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid) receptor.
Glutamate activates the ion channels in both NMDA and AMPA receptors. When glutamate binds NMDA receptors, they open up to allow calcium ions into the cell. When glutamate binds AMPA receptors, they open up to allow sodium ions into, and potassium ions out of, the cell. No matter what type of ion enters the cell, the neuron is depolarized and the action potential “jumps” from the presynaptic cell to the postsynaptic cell. What happens after the propagation of the action potential depends on whether the neuron inhibits or excites downstream targets and the function of the neural circuit it’s in.
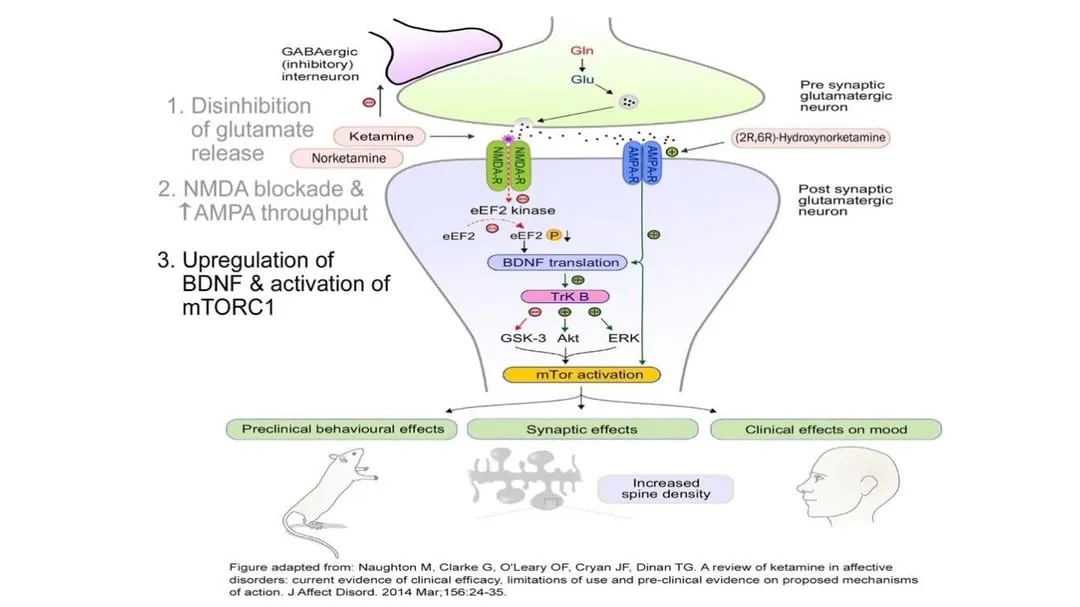
At the very low dose shown to have an antidepressant effect, ketamine appears to increase the release of glutamate from the presynaptic neuron into the synaptic cleft. The ketamine then preferentially blocks glutamate at the NMDA receptors of the postsynaptic cell but doesn’t block glutamate binding to adjacent AMPA receptors. The net effect is to increase AMPA activation. The effect is magnified by the way ketamine induces the neuron to make additional AMPA receptors and move them into the membrane of the synapse area.
Fertilizing the Brain
By increasing the level of glutamate transmission while also shifting the balance of glutamate activation from NMDA to AMPA receptors, ketamine rapidly upregulates neuronal production and release of BDNF (brain-derived neurotrophic factor). Aptly called fertilizer for the brain, BDNF is a protein that helps promote the growth, maintenance, and survival of neurons—in other words, it enhances neuroplasticity.
At the same time, ketamine stimulates a central cell pathway called mTOR (mammalian target of rapamycin), which regulates many processes involved in cell growth, including synthesizing the proteins needed for long-term memory. In combination with increased BDNF production, mTOR stimulation improves synaptic connectivity in the prefrontal cortex and hippocampus, key areas of the brain associated with emotional regulation, and reverses the synaptic damage that occurs in these areas when the brain is subjected to chronic stress. Owing to these neuroplastic effects, regrowth of dendritic spines can happen within a few hours of a therapeutic ketamine dose. When the atrophied neurons can repair the damage and regrow their connections with other neurons, symptoms of depression and anxiety improve.
At higher doses, ketamine appears to lose its discriminating effect and instead blocks both NMDA and AMPA receptors. When both receptor types are blocked, upregulation of BDNF and enhanced neuroplasticity stops. This could explain why it took so long to recognize the antidepressant effect of ketamine. It has been used as a surgical anesthetic since the 1970s, but it was only when researchers at Yale started using it in much lower doses as a psychiatric treatment that the antidepressant effect was serendipitously discovered.
Other Mechanisms
Evolving research is revealing more about the mechanism of action for ketamine. One additional potential mechanism for ketamine is the NMDA receptors on a type of neuron called the parvalbumin interneuron. These neurons are small and relatively rare in the brain, but they’re key to synchronizing electrical activity in the brain. They work by inhibiting activity in the neurons around them. When a subunit of the NMDA receptors on these neurons is blocked by ketamine, the cells don’t get as much glutamate, and their inhibitory effect is reduced. The parvalbumin interneurons act like a brake on brain activity. Ease up on the brake and all the brain circuits become more active. The brain wakes up and becomes more responsive.
While ketamine binds strongly to NMDA and AMPA receptors, it’s also taken up, to a much smaller degree, by other neuroreceptors. Ketamine can trigger increased release of the neuromodulators dopamine and noradrenaline; it also binds weakly to nicotinic and opioid receptors. Exactly what role these receptors play in the antidepressant effect is still largely unknown and even contradictory. For example, some studies seem to show that ketamine binds to opiate receptors in a way that could create addiction; other studies show that the potential for addiction is very low.
Another possible mechanism for ketamine is its action on structures within the neuron. Some studies suggest that ketamine accumulates in lysosomes (organelles that contains digestive enzymes) and synaptic vesicles, which might then trigger mTOR signaling. Ketamine could also affect the endoplasmic reticulum (tubules within the cell that are involved with protein and lipid synthesis) and the Golgi bodies (organelles that sort, process, and transport proteins). The mechanisms are complex and still being unraveled.
Ketamine and Connectivity
The impacts of stress and depression on the brain aren’t just at the molecular and cellular level. Depression impacts connectivity at the brain macro level as well. By using functional magnetic resonance imaging (fMRI), researchers have been able to see how people with depression seem to have weaker connections within larger neural networks, such as the prefrontal cortex. This portion of the brain is the seat of higher-level cognitive processes, including executive function —the ability to control short-term behaviors in favor of self control, planning, decision making, problem solving, and long-term goals. When the subregions of the prefrontal cortex aren’t communicating well, as is the case in depression, executive function can be dysfunctional. Ketamine seems to help improve global connectivity in this portion of the brain and improve the linkages among the subregions.
References
- Abdallah, C. G., Dutta, A., Averill, C. L., McKie, S., Akiki, T. J., Averill, L. A., & Deakin, J. (2018). Ketamine, but not the NMDAR antagonist lanicemine, increases prefrontal global connectivity in depressed patients. Chronic stress (Thousand Oaks, Calif.), 2.
- Berman, R.M., Cappiello, A., Anand, A., Oren, D.A., Heninger, G.R., Charney, D.S. and Krystal, J.H., 2000. Antidepressant effects of ketamine in depressed patients. Biological psychiatry, 47(4), pp.351-354.
- Duman, R.S. and Aghajanian, G.K., 2012. Synaptic dysfunction in depression: potential therapeutic targets. Science, 338(6103), pp.68-72.
- Fava, M., Freeman, M.P., Flynn, M., Judge, H., Hoeppner, B.B., Cusin, C., Ionescu, D.F., Mathew, S.J., Chang, L.C., Iosifescu, D.V. and Murrough, J., 2018. Double-blind, placebo-controlled, dose-ranging trial of intravenous ketamine as adjunctive therapy in treatment-resistant depression (TRD). Molecular psychiatry, p.1.
- Li, N., Lee, B., Liu, R.J., Banasr, M., Dwyer, J.M., Iwata, M., Li, X.Y., Aghajanian, G. and Duman, R.S., 2010. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science, 329(5994), pp.959-964.
- Luckenbaugh, D.A., Niciu, M.J., Ionescu, D.F., Nolan, N.M., Richards, E.M., Brutsche, N.E., Guevara, S. and Zarate, C.A., 2014. Do the dissociative side effects of ketamine mediate its antidepressant effects?. Journal of affective disorders, 159, pp.56-61.
- Murrough, J.W., Perez, A.M., Pillemer, S., Stern, J., Parides, M.K., aan het Rot, M., Collins, K.A., Mathew, S.J., Charney, D.S. & Iosifescu, D.V. (2013). Rapid and longer-term antidepressant effects of repeated ketamine infusions in treatment-resistant major depression. Biological psychiatry, 74(4), pp.250-256.
- Lester, H. A., Miwa, J. M., & Srinivasan, R. (2012). Psychiatric drugs bind to classical targets within early exocytotic pathways: therapeutic effects. Biological psychiatry, 72(11), 907–915. doi:10.1016/j.biopsych.2012.05.020
- Sleigh, J. et al. (2014). Ketamine–More mechanisms of action than just NMDA blockade. Trends in anaesthesia and critical care, 4 (2-3), 76–81. doi.org/10.1016/j.tacc.2014.03.002.
- Perry EB Jr, Krystal JH; Yale Ketamine Study Group, et al. Psychiatric safety of ketamine in psychopharmacology research. Psychopharmacology (Berl). 2007 Jun;192(2):253-60.